MCMC Diagnostics in R with the coda Package
This is a follow up to my recent post introducing the use of JAGS in R through the rjags package. In the comments on that post, Bernd Weiss encouraged me to write a short addendum that describes diagnostic functions that you should use to assess the output from an MCMC sampler.
I’ve only been using these diagnostics for a week now for an academic project of my own, so I’ll summarize my understanding of their use as it stands today. Please correct me if I’m spreading misinformation.
As I see it, all diagnostics used to analyze the output of an MCMC sampler try to answer a simple question: has the sampler been given a sufficient adaptation (“burn-in”) period to justify your claim that the samples you draw from the chains approximate the posterior distribution of interest to you? This question in turn leads one to analyzing the samples drawn after the burn-in period for obvious warning signs. To test for these potential warning signs, it’s easiest to use the diagnostic functions that are included in coda.
To make the use of coda clear, I’m going to follow up on the linear regression example that I used in my last post. First, we generate some data from a linear model:
library('rjags')
N <- 1000
x <- 1:N
epsilon <- rnorm(N, , 1)
y <- x + epsilon
Then we set up our BUGS/JAGS model in example.bug:
model {
for (i in 1:N) {
y[i] ~ dnorm(y.hat[i], tau)
y.hat[i] <- a + b * x[i]
}
a ~ dnorm(0, 0.0001)
b ~ dnorm(0, 0.0001)
tau <- pow(sigma, -2)
sigma ~ dunif(0, 100)
}
All of this is copied exactly from my earlier post. Now we change things by replacing our call to jags.samples() with a call to coda.samples(). This will provide output from JAGS in the format necessary for using the two diagnostic functions I understand best: plot() and gelman.plot().
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
n.chains = 4,
n.adapt = 10
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_1.png')
plot(samples)
dev.off()
Because we have used such a small number of adaptive samples (only 10), our call to jags.model will produce this warning message:
#Warning message:
#In adapt(model, n.adapt) :
# Adaptation incomplete. Recreate the model with a longer adaptive phase.
Thankfully, this message is not the only evidence that we’ve used too few adaptive samples: you can also tell from the output of our call to plot():
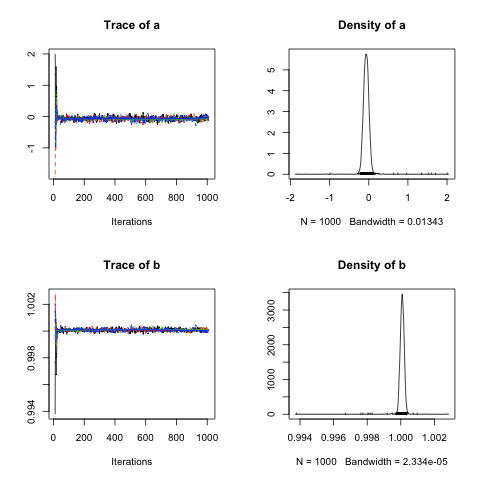
The density plots for a and b in the right column are very suspicious: they are extremely pointed and seem to include a large number of outlier values that don’t belong if the values for a and b are normally distributed. You can also see these extreme values in the trace plots in the left column as well: they are the extreme values at the start of the trace for each chain.
Simply using 1,000 adaptive samples instead of 10 makes a world of difference:
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
n.chains = 4,
n.adapt = 1000
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_2.png')
plot(samples)
dev.off()
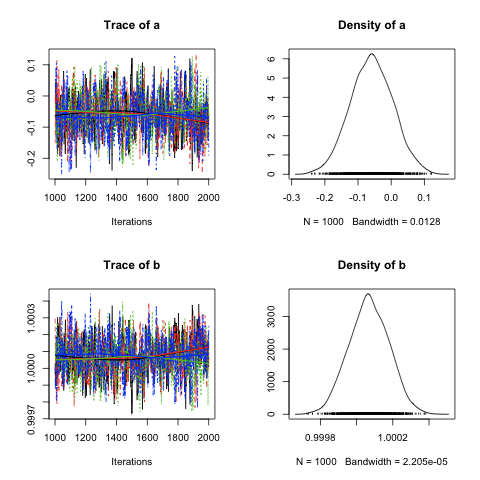
In this image, you can see that the trajectory of the chain is consistent over time and that its distribution looks appropriately normal. So the first takeaway message is simple: check the traces and distributions of your variables using plot() to make sure that they are reasonable and don’t indicate clear deficiencies in the length of your adaptation period. When you know the distributional form of your posteriors, this is particularly effective, as in our example here, where we know to expect normal distributions.
Another diagnostic tool is the Gelman plot, which has a simple logic to it: you test how similar the various chains you’re using are as a way to detect whether they’ve hit the target distribution. This similarity is what’s called mixing. To start, it’s easier to simply repeat our examples above using the Gelman plot:
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
n.chains = 4,
n.adapt = 10
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_3.png')
gelman.plot(samples)
dev.off()
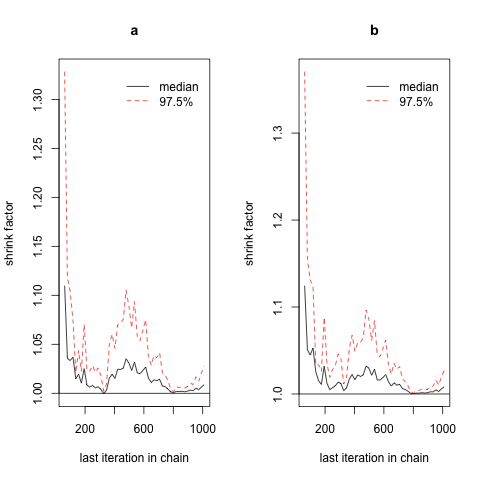
And then you can try to see how the plot looks different when you switch to a longer adaptive period:
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
n.chains = 4,
n.adapt = 1000
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_4.png')
gelman.plot(samples)
dev.off()
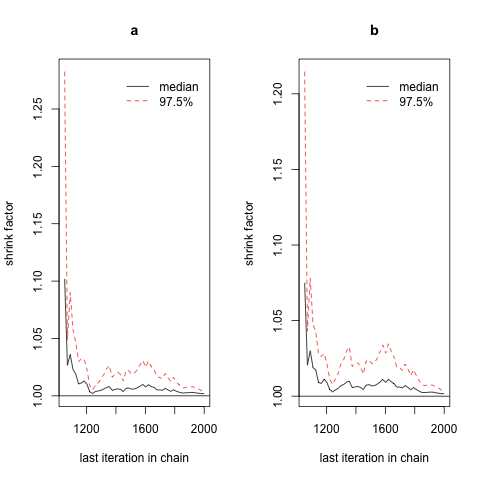
Unfortunately, given our current call to jags.model(), it’s quite hard visually to identify convergence using Gelman plots, since the scales of these plots are not identical across our two examples, and the most prominent visual patterns are likely to be the results of random noise. There is a reason for this difficulty: we’re not properly initializing our sampler’s starting values separately for each chain. Both chains start from identical positions, which means that we don’t have enough power to really see the size of the space a theoretical chain might pass through before settling down. To fix that, we change our call to jags.model() to include an inits value, for which we provide an anonymous function that provides random values consistent with the prior we specified in example.bug. First, let’s repeat our previous approach again:
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
n.chains = 4,
n.adapt = 10
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_5.png')
gelman.plot(samples)
dev.off()
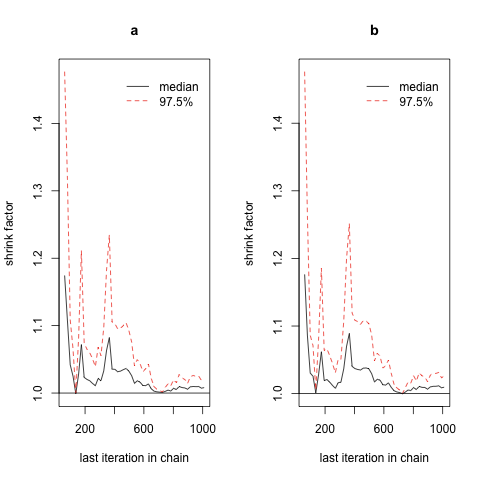
Now let’s add proper random initialization for the starting position of each chain:
jags <- jags.model(
'example.bug',
data = list(
'x' = x,
'y' = y,
'N' = N
),
inits = function () {
list(
'a' = rnorm(1, 0, 100),
'b' = rnorm(1, 0, 100)
)
},
n.chains = 4,
n.adapt = 10
)
samples <- coda.samples(
jags,
c('a', 'b'),
1000
)
png('plot_6.png')
gelman.plot(samples)
dev.off()
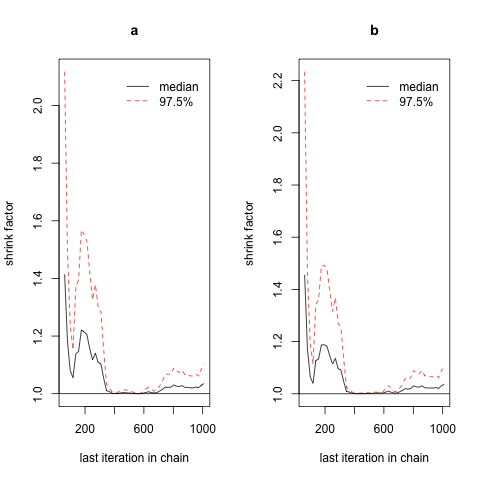
Now you should be able to easily see that the chains are converging near the end of the sampling period, and that we would do well to give ourselves a greater adaptation period before using any of the samples we’ve generated, since the early samples in the chain are far too dispersed.
With these two techniques you should be able to diagnose obvious deficiencies in the length of your adaptive period.